Diesen Nachweis haben jetzt Wissenschaftler vom Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) und vom Max-Delbrück-Centrum für Molekulare Medizin (MDC) zusammen mit einem internationalen Forscherteam erbracht, nachdem bei drei betroffenen Kindern die gleiche Punktmutation gefunden wurde. ClC-6 ist einer von neun Mitgliedern der CLCN Genfamilie von Chloridkanälen und Chlorid/Protonen-Austauschern und war neben ClC-3 bislang der einzige, dem noch keine menschliche Erkrankung zugeordnet werden konnte. Die Ergebnisse wurden soeben im American Journal of Human Genetics publiziert.
Unter dem Begriff „lysosomale Speicherkrankheit“ werden eine Reihe von genetisch bedingten Stoffwechselerkrankungen zusammengefasst, die auf eine fehlerhafte oder unzureichende Funktion der Lysosomen zurückzuführen sind. Die Zellorganellen spielen sowohl bei der zellulären Müllabfuhr, als auch bei der Regulation des zellulären Stoffwechsels eine wichtige Rolle. Ist ihre Funktion gestört, reichern sich in den betroffenen Zellen oft Substanzen an, die der Körper nicht mehr benötigt – mit zum Teil fatalen Folgen für Körper und Geist. Häufig sind Zellen des zentralen Nervensystems betroffen, was zu einer mehr oder minder ausgeprägten Neurodegeneration führt.
Die Ursache für eine besonders schwere Form einer genetisch bedingten Neurodegeneration konnten Forscher vom Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) und dem Max-Delbrück-Centrum für Molekulare Medizin (MDC) jetzt zusammen mit Kooperationspartnern aus Hamburg, Rom und den USA nachweisen: Es ist eine Mutation im CLCN6-Gen, die bei drei nicht miteinander verwandten Kindern aus Italien, Deutschland und den USA zu erheblichen Entwicklungsstörungen führt, zusammen mit geistiger Retardierung, verringertem Muskeltonus, Atemschwierigkeit, Sehstörungen und nachweislichem Verlust von Hirnsubstanz.
Ionentransporter ClC-6 gehört zur Chloridkanalfamilie
Humangenetiker, darunter der Co-Studienleiter Marco Tartaglia aus Rom und Kerstin Kutsche aus Hamburg, hatten unabhängig voneinander die gleiche Punktmutation bei ihren kleinen Patienten entdeckt und das Team von Prof. Thomas Jentsch gebeten, Effekte der Mutation auf den Ionentransporter und seine zelluläre Funktionen zu überprüfen . Als Entdecker der CLC Chloridkanalfamilie hatte Jentsch schon bei fast allen neun CLC-Genen verschiedene Mutationen gefunden oder charakterisiert, die unterschiedlichste Erbkrankheiten verursachen können. Nur den Ionentransportern ClC-3 und ClC-6 konnten bisher noch keine menschlichen Erkrankungen zugeordnet werden. „Wir hatten schon vor fünfzehn Jahren ClC-6 Knockout-Mausmodell hergestellt, das eine leichte lysosomale Speicherkrankheit aufwies, konnten aber keinen Patienten mit einer ähnlichen Funktionsverlustmutante finden“, erklärt Prof. Jentsch. „Jetzt haben wir eine anders geartete CLCN6 Mutation bei einer wesentlich schwereren Erkrankung des Menschen gefunden.“
Schon das Vorkommen der exakt gleichen Mutation bei drei unabhängigen Patienten mit dem gleichen Krankheitsbild deutet auf eine kausale Rolle der Mutation hin, aber erst die funktionelle Analyse in Zellkultur brachte endgültige Gewissheit und führte zur Klassifizierung als lysosomale Erkrankung. „Mit an Sicherheit grenzender Wahrscheinlichkeit führt der von uns nachgewiesene Funktionsgewinn der ClC-6 Mutante zu diesem besonders schweren Krankheitsbild, dem eindeutig eine Störung in Lysosomen zugrunde liegt. Deshalb gehen wir zusammen mit unseren Kooperationspartnern von einer lysosomalen Speicherkrankheit aus“, betont Thomas Jentsch. Ein endgültiger Beweis für diese Klassifizierung jedoch eine post mortem Untersuchung von Hirnschnitten von Patienten oder einem neuen Mausmodell, das die gleiche Mutation trägt, erfordern.
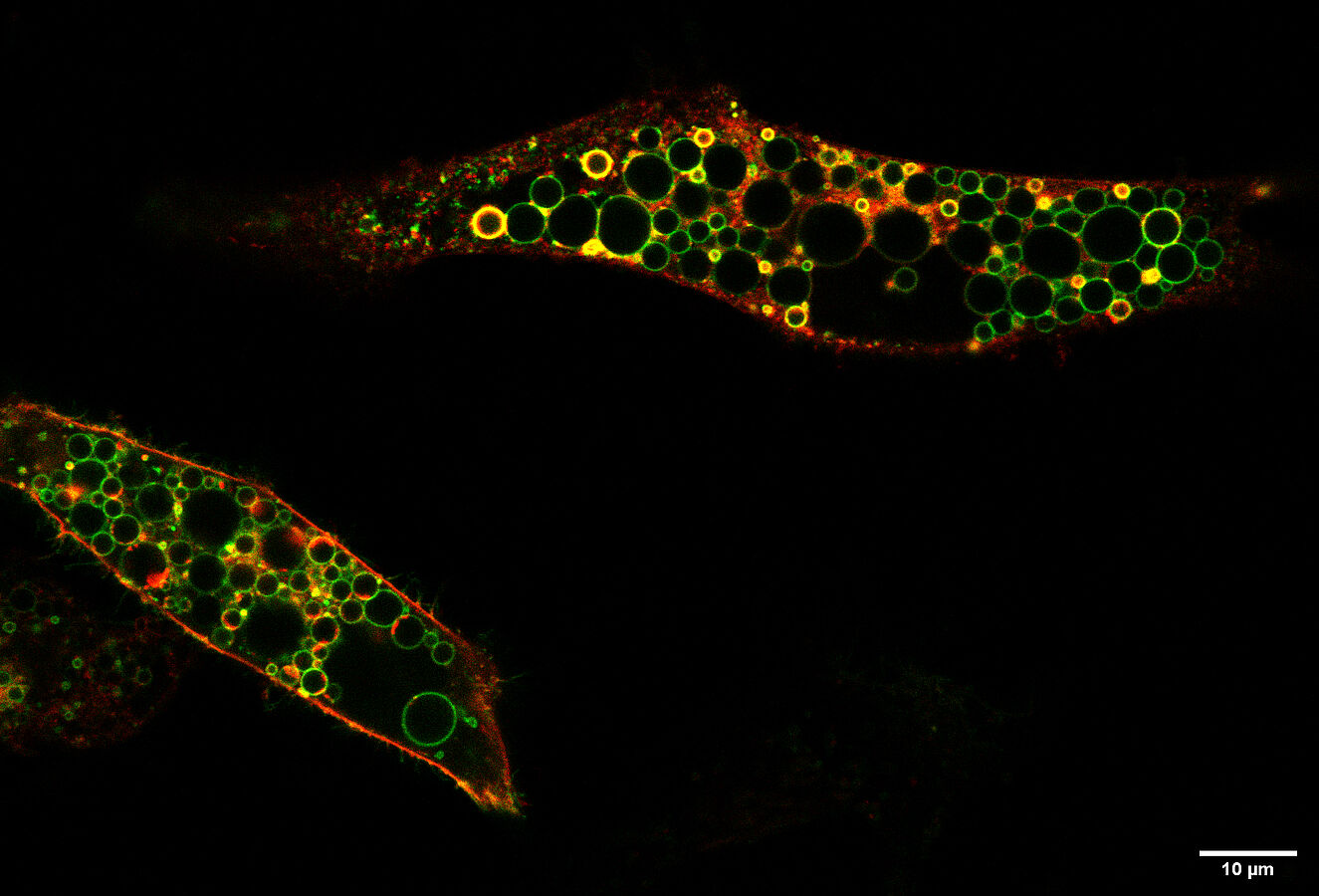
Mehr Chloridaufnahme führt zu abnormal großen, Lysosom-ähnlichen Vesikeln
Anders als die Chloridkanäle ClC-1, -2, -3 und -K sitzen die Chlorid/Protonen-Austauscher ClC-3, -4, -5, -6, und -7 nicht auf der Plasmamembran, sondern in intrazellulären Membranen, hauptsächlich auf Endosomen und Lysosomen. Jentsch und sein Team hatten vor einiger Zeit bereits Mutationen von ClC-7 als Ursache für eine mit Osteopetrose assoziierten Form der lysosomalen Speicherkrankheit ausmachen können und Mutationen von ClC-4 führen u.a. zu intellektuellen Defiziten. Während ClC-7 fast rein lysosomal ist, sitzt ClC-6 vorwiegend auf späten Endosomen – einer Art Vorstufe von Lysosomen.
Die Untersuchungen der Berliner Forscher zeigten einen hyperaktiven Ionentransporter ClC-6, dessen Transport von Chlorid und Protonen stark erhöht ist und der nicht mehr pH-sensitiv ist. Ein saures pH-Milieu, wie es beim graduellen Übergang von Endosomen zu Lysosomen erreicht wird, schaltet den Transporter normalerweise ab. Diese Regulation fehlt bei der kankheitserzeugenden Mutante. Durch den erhöhten, unregulierten Ionentransport – ein pathologischer Funktionsgewinn – entstanden in Zellen, in die die Forscher den mutierten ClC-6 einbrachten, drastisch vergrößerte, Lysosomen-ähnliche Vesikel. Jentsch zufolge kann dieser Funktionsgewinn die Krankheit der Kinder recht gut erklären. „Vesikel, auf deren Membran das mutierte ClC-6 sitzt, werden durch gesteigerte Aufnahme von Chlorid und später Wasser pathologisch vergrößert. Diese Aufnahme wird durch den ClC-6-vermittelten Austausch von Protonen angetrieben, die im sauren Milieu von Endosomen und Lysosomen reichlich vorhanden sind. Dies führt zu einer Einschränkung der Funktion von Lysosomen und langfristig wohl zu lysosomaler Speicherung in Nervenzellen, die sich ja nicht mehr teilen können. Zusätzlich führt die Gewebsverteilung von ClC-6, der fast ausschließlich in Nervenzellen vorkommt, zu dem vorwiegend neurologischen Krankheitsbild“
„Die vorliegende Arbeit unterstreicht, wie wichtig der Ionentransport für den endosomal-lysosomalen Weg ist“, sagt Jentsch. „Wir sehen ein großes Spektrum von Erbkrankheiten, die durch Mutationen an diesen vesikulären CLCs sowie anderen intrazellulären Kanälen verursacht werden können.“ Ganz unterschiedliche Organe können betroffen sein: Zum Beispiel führen Mutationen im endosomalen ClC-5 zu Nierensteinen und Proteinverlust in den Urin, wie Jentsch und Kollegen bereits vor Jahren zeigen konnten.
Jentsch ist zuversichtlich, auch bald den ClC-3 Austauscher mit einer Erbkrankheit in Verbindung zu bringen – eine vor Jahren von der Gruppe publizierte KO Maus zeigt dramatische Neurodegenerationen. Zusammen mit dem jetzigen Befund würden damit alle neun CLCN Gene als Krankheitsgene des Menschen entlarvt. „Die Lücke schließt sich“, sagt Jentsch, „und wir sehen hieran sehr schön, welche Bedeutung die Grundlagenforschung – der erste CLC wurde von uns aus einem elektrischen Fisch kloniert - für die Diagnose und das Verstehen von Erkrankungen hat.“
